Document Type : Original Article
Authors
- Md Abul Hasan Roni 1
- Md. Golam Mortuza 2
- , Rozina 3
- Ripon Kumar Shaha 4
- Sajidul Hoque 5
- Ajoy Kumer 6, 7
1 Faculty of Chemical and Process Engineering Technology, Universiti Malaysia Pahang Al-Sultan Abdullah (UMPSA), Gambang, Malaysia
2 Department of Science & Humanities, Bangladesh Army International University of Science and Technology, Cumilla, Bangladesh
3 Department of Gynecology and Obstetrics, Mainamoti Medical College and Hospital, Cumilla
4 Department of Chemistry, Sreepur Govt. Degree College, Sreepur, Magura, Bangladesh
5 Department of Pharmaceutical Sciences, North South University Bashundhara, Dhaka, Bangladesh
6 Laboratory of Computational Research for Drug Design and Material Science, Department of Chemistry, College of Arts and Sciences, IUBAT—International University of Business Agriculture and Technology, Uttara Model Town, Dhaka, Bangladesh
7 Center for Global Health Research, Saveetha Institute of Medical and Technical Sciences in Saveetha Medical College and Hospital, Chennai, India
Abstract
Having a wide range of pharmacological action antiviral properties of alkaloids, these were focused on this study for computational screening against SARS-CoV-2 proteases. However, 256 alkaloids from PubChem databased, 10 compounds were selected by PASS prediction for primally observation having active value against virus with the main protease (Mpro) of acute respiratory syndrome coronavirus-2 (SARS-CoV-2). Besides, the pharmacokinetics and Lipinski's rule of five (RO5) were evaluated for all molecules being drug likeness properties, and then these were selected for molecular docking against the Mpro of corona virus, and L-03, L-07, and L-13 found the best-bonded molecules accounting weak H bonding and hydrophobic bond interactions. In addition, on the binding energies of Mpro, L-3, L-7, L-13, L-18, L-25, L-26, L-34, L-39, L-42, and L-49 were at -8.6, -8.3, -8.2, -8.1, -8.0, -7.9, -7.9, -7.9, and -7.9 kcal/mol. Overall, L-3 is considerable best candidate with Mpro in view of the molecular docking score. In addition, the quantum properties, chemical hardness and softness values were calculated. Finally, from admetSAR online database, absorption, distribution, metabolism, excretion, and toxicity (ADMET) were evaluated. Lastly, L-3, L-7, and L-13 were satisfied the toxicity category. In comparison to the World Health Organization (WHO) guidelines, FDA-approved antiviral drugs, favipiravir and remdesivir, which have binding energies of -5.1 and -8.1 kcal/mol of the Mpro, respectively. In this case, our selected L-3, L-7, L-13, and L-18 have binding energies of -8.6, -8.3, -8.2, and -8.2 kcal/mol, respectively. Thus, the L-3, L-7, L-13 and L-18 molecules will have strong activity against the SARS-CoV-2.
Graphical Abstract
)
Keywords
Main Subjects
Introduction
SARS-CoV-2 infected people for the first time in 2019 in China, and spread out over the world in short time which was the record breaking mobility was that no other epidemic could be able to cover the whole world as the capacity of SARS-CoV-2 in previous any time [1-3]. Initially, the large family of single-stranded RNA viruses also crown-shaped RNA viruses, causes to infection for animals and humans, triggering respiratory, gastrointestinal, hepatic, and neurologic diseases [4]. SARS-CoV-2 is pleomorphic, but spherical or elliptic with a diameter of 60-140 nm. Its 29,891-nucleotide RNA genome encodes 9,860 amino acids and shares 99.9% homologs with the bat genome (bat-SL-CoVZC45), revealing a recent host transfer into humans [5]. This Mpro is one of the crucial targets for combating the recent pandemic due to SARS-CoV-2 [6-8]. The virus spreads through saliva, coughing, sneezing, and close personal contact to human being [9]. Inflammatory cytokines are produced by coronaviruses which is caused to damage of organ by a "cytokine cascade" or "cytokine storm" [10]. Still, the major fact for SARS-CoV-2 is that we do not invent any effective drug for treatment although some antiviral drugs like favipiravir (D-1), remdesivir, hydroxy-chloroquine, azithromycin, lopinavir/ritonavir, and nafamostat mesylate are being tested and recommended as a treatment in clinical trials, but not directly established for treatment [11-12]. Novel influenza virus treatment is approved in Japan and China with favipiravir [13]. It is also effective against Ebola and RNA viruses. Favipiravir is recommended for COVID-19 treatment by the world health organization (WHO) and the national institutes of health (NIH) [14] and the FDA has approved remdesivir (D-2) for adults and certain pediatric patients [15].
Alkaloids are a miscellaneous group of natural phytochemicals. These phytochemicals in plants afford them protection against pests, and herbivorous organisms and also regulator their progress. Plentiful of these alkaloids could be used in various biological effects, even developed into medications with different medicinal properties [16]. It is considered a broad group of naturally happening organic compounds, forming most of the largest group of phytochemicals, where the nitrogen atoms are a key distinctive trait of alkaloids consequential in their alkaline properties and medicinal actions. Amino acids for example tyrosine, lysine, ornithine, phenylalanine, and tryptophan are used to synthesize most of alkaloids. Fortunately, the most common plant-derived antiviral agents are alkaloids [17-18]. The largest group of plant secondary metabolites include a vast range of naturally occurring chemical substances [19]. Natural alkaloids are antioxidant, anti-inflammatory, and anti-viral [20-21]. In addition, alkaloids contain many promising anti-coronavirus compounds [22] which are very available in our natural resources, such as coffee, chocolate, tea, tomatoes, and potatoes [23]. They are also commonly used to treat neurological disorders, cancer, metabolic problems, and infectious diseases. The hepatitis B virus was tested against alkaloids from Corydalis saxicola Bunting (Papaveraceae) [24].
To recall, having the wide range of antiviral medicinal properties, it is urgent need to make a profile among all of listed alkaloids to estimate their activity. However, we are trying to inhibit SARS-protease CoV-2's (Mpro) (PDB ID: 6M03, Resolution: 2.00 Å, Total Structure Weight: 33.83 kDa, Deposited Residue Count: 306, Unique protein chains: 1) using computational approaches like molecular docking, PASS prediction, and ADMET properties.
Materials and methods
Pass Prediction
Using the Prediction of Activity Spectra for Substances (PASS)-Way2Drug server (http://www.pharmaexpert.ru/passonline/), the biological activities of the selected alkaloids were determined. With Pa and Pi values, the possibly active or inactive ligands were predicted. The proposed active chemical should have Pa values near 1 and Pi values near zero [25].
Ligand preparation and optimization
We chose 50 alkaloid-containing compounds from the PubChem database. PyRx 0.8 was used to generate the 3D structures of the ligands [26]. The energy form of all compounds was reduced and translated into pdbqt format using Open Babel in PyRx version 0.8. Molecular optimization was performed utilizing the DFT functional method. While an electronegative atom, oxygen, appeared to be present, the functional, B3LYP, and basis sets, DNP+, were employed for function setup in the DMol3 code to provide extremely precise results. After improving the analytical processes, the border molecular orbitals (HOMO and LUMO) and magnitudes of HOMO and LUMO were developed. The optimized chemical compounds were saved as a pdb file in preparation for future computational work such as molecular docking and ADMET calculations [27].
Protein preparation
The crystal structure of SARS-CoV-2 Mpro (PDB ID: 6M03, Resolution: 2.00 Å) was retrieved from the RCSB Protein Data Bank (http://www.rcsb.org) [28] using the protein preparation wizard of PyMol version 1.1.0 to prepare the modeled Mpro for docking analysis by removing water molecule. Open Babel was used to convert the file into pdbqt [29].
Molecular docking
For docking studies on viral protein with ligand, we used Auto Dock Vina with a grid box radius of 10.0 around the active site region. The grid center points were set to X = 12.11, Y = -11.38, Z= 4.66, and the dimension (Å) X = 36.82, Y = 64, Z = 62.07 of Mpro. The gid box wrapped the protein's substrate-binding domain. DS visualizer illustrated Auto Dock Vina's performance [29].
Pharmacokinetics properties analysis
Swiss ADME and pkCSM-pharmacokinetics software are used for the prediction pharmacokinetics of listed molecules [25,30]. ADMET, solubility, carcinogenicity, bioavailability, and drug-likeliness property were studied in this part [31].
Results and Discussion
Prediction of biological activities
All compounds' bioactivities were assessed using the PASS online program. Lipinski's rule of five evaluated 50 chemicals (RO5). Forty-two showed the necessary antiviral activity, with Pa > Pi (Supplementary: ST-1), and the top 10 molecules were chosen based on docking score, as listed in Table 1. FDA-approved antiviral drugs favipiravir and remdesivir have Pa values of 0.447 and 0.814, whereas the L-3, L-7, L-18, L-25, and L-34 Pa values have shown 0.508, 0.335, 0.219, 0.450, and 0.409. Therefore, these values indicate that the L-3, L-7, L-18, L-25, and L-34 candidate molecules may have strong activity against the MPro (6M03).
Molecular docking analysis
A total of 50 dockings of alkaloids were performed against the main protease protein of MPro (6M03). The highest docking score was -8.6 kcal/mol (L-3), and the lowest score was -5.5 kcal/mol (L-49). Forty-two alkaloids had docking scores greater than 6 (Supplementary: ST-2). Out of 42, we got six compounds with docking results ranging more than or equal to -8.00 kcal/mol. Four compounds rang at -7.9 kcal/mol, 23 compounds rang at more than or equal to -7 kcal/mol, and eight were more than or equal to -6.0 kcal/mol (Supplementary: ST-2). Interestingly, ten bioactive alkaloids out of 42 have a docking score of -8.6 to -7.9 kcal/mol. Six compounds have a docking score greater than -8.0 kcal/mol, and four compounds have a score of -7.9 kcal/mol.
Binding energy is about -5.1 and -8.1 kcal/mol for the approved antiviral drugs favipiravir and remdesivir. Whereas the selected ligands are showing strong binding affinity compared to standard antivirals. Based on the results of binding affinity, it is observed that L-3, L-7, and L-13 have better binding affinity against the MPro(6M03).
L03 is called Densiflorine, a type of C20 alkaloid that can be found in various plant species. Alkaloids are organic compounds that often have pharmacological effects and are commonly found in plants. Densiflorine may be present in certain plant families, such as the Apocynaceae and Rubiaceae families, which are known to contain alkaloids. Some plants within these families are known to produce alkaloids, including species like Vinca (Catharanthus roseus), Rauwolfia (Rauvolfia serpentina), and Tabernaemontana (Tabernaemontana spp.). L07 is compound of Catharanthine, an alkaloid found in the Madagascar periwinkle plant (Catharanthus roseus), also known as the vinca or rosy periwinkle. This plant is native to Madagascar, but is now cultivated in various parts of the world. Catharanthine is primarily sourced from the leaves and stems of the Madagascar periwinkle plant. This plant is well-known for its medicinal properties and is cultivated for the production of various alkaloids, including catharanthine and vincristine. Catharanthine is one of several alkaloids present in the plant, and it has both pharmaceutical and ecological significance.
L13 is commonly called as Brucine, often referred to as Brucinum in Latin, is a bitter alkaloid found in certain plants of the genus Strychnos, particularly Strychnos nux-vomica and Strychnos ignatii. It is structurally related to strychnine, another alkaloid found in the same plants. Brucine is known for its extreme bitterness. Brucine is primarily obtained from the seeds of Strychnos nux-vomica and Strychnos ignatii, commonly known as nux vomica and Saint Ignatius' bean, respectively.
Table 1. Ligand No, PubChem CID, and Pa>Pi values of top 10 molecules

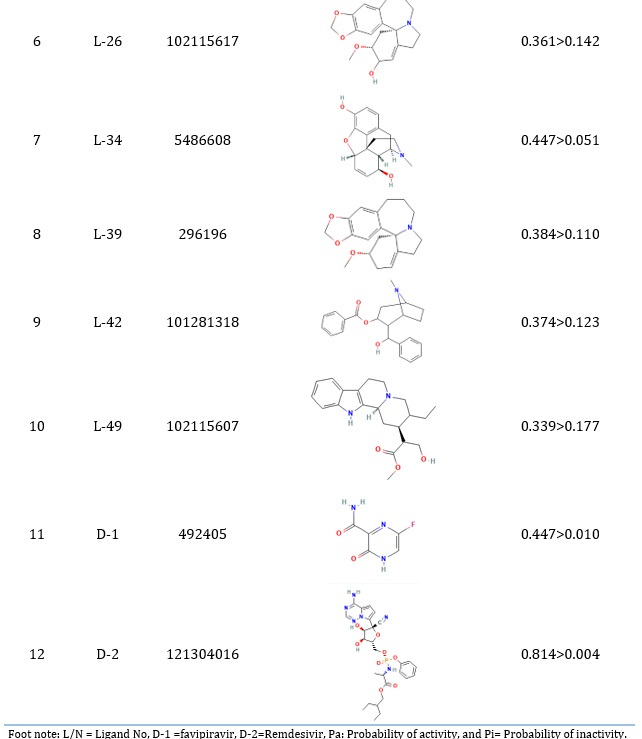
Table 2. Best molecular docking scores (kcal/mol) against main protease (Mpro) of SARS-CoV-2 (PDB ID: 6M03)

Redocking can serve as a form of validation for docking studies by assessing the ability of a molecular docking protocol to accurately reproduce known binding poses of ligands within a protein's binding site. Here is how re-docking can perform validation of docking. The ligand is extracted from the crystallographic complex, and the protein structure is prepared (e.g., energy minimization, addition of missing atoms). The same molecular docking software and parameters used for virtual screening or pose prediction are applied to dock the ligand back into the protein's binding site. RMSD measures the average distance between the heavy atoms of the ligand in the redocked pose and the crystallographic pose. A low RMSD value indicates a close match between two poses, suggesting that the docking protocol is capable of accurately predicting the binding pose. Therefore, each molecular docking, the RMSD can be calculated at 0 A that indicates the docking validation.
Protein-ligand interaction
In this work, 50 bioactive alkaloid compounds were chosen for further examination because they displayed a binding affinity of -5.0 kcal/mol or higher for the target protein, and may all be employed as lead compounds. Hydrogen bonds and hydrophobic bonds (non-bond interactions) with residues were utilized, whereas the remaining interactions were avoided. Moreover, the bond distance is determined here. All associated information and 2D images of particular compounds (L-3, L-7, L-13, L-18, L-25, L-26, L-34, L-39, L-42, and L-49) were obtained (Supplementary: ST-3).
In L-3, there is no hydrogen bond. It has two hydrophobic bonds, which are residues PHE 294 (3.70 A°), ARG 298 (5.05 A°). L-7 contains one hydrogen bond with GLN110 (2.12A°) and two hydrophobic bond with VAL 303 (3.87 A°) and ARG 298 (4.10 Å). L-13 interacts with the MPro(6M03) via three hydrophobic bond interactions involving the residues ARG298 (5.38 Å), VAL303 (4.47 Å) and PHE 294 (4.03Å), two of which are conventional hydrogen bonds and one of which is a carbon-hydrogen bond. Two hydrogen bonds involved residues GLN110 (2.17 Å) and PHE 294 (4.13 Å). L-18 and the MPro(6M03) complex are composed of hydrogen and hydrophobic interactions, with residues GLN 110 (2.48 A°), ASP295 (3.69 Å) and VAL104 (4.61 Å) and THR111 (4.10 Å). In L-25, there are no present any hydrogen bonds. It has three hydrophobic bonds, which are residues PHE294 (4.59Å), THR292 (1.41 Å) and ARG298 (4.38 Å). L-26 has one hydrogen bond and two hydrophobic interactions spread on residues GLN110, ARG298 (4.71 Å), and PHE294 (4.40 Å).
L-34 shows two kinds of interactions that were predicted between L-34 and the main protease 6M03. One is a hydrogen bond, and another is a hydrophobic bond, engaged respectively with THR292 (3.09 Å) and ASP295 (2.73 Å), PHE294 (4.32 Å), PHE 305 (5.48 Å). L-39 has one hydrogen bond and two hydrophobic bond interactions with GLN110 (2.10), ARG298 (4.54Å), and VAL303 (3.87Å). L-42 binds to MPro(6M03) via three hydrogen bonds and four hydrophobic interactions with the following residues: ASN151 (2.91 A°), THR 292 (2.14 Å), ILE 152 (3.18 Å), PHE294 (4.18 Å), ARG298 (4.59 Å), VAL303 (4.37 Å), and THR 111 (2.50 A°). Two kinds of interactions were predicted between L-49 and the main protease MPro(6M03). It accounts for the two H-bonds and two hydrophobic interactions engaged, respectively, with THR292 (2.29 Å), THR111 (3.45 Å), PHE294 (4.28 Å), and ARG298 (5.21 Å).
Favipiravir (D1) binds to the MPro (6M03) via four hydrogen bonds and only one hydrophobic interaction with the following residues: THR292 (2.37 Å), ASN151 (2.07 Å), THR111 (2.92 Å), ILE152 (2.92 Å), and PHE8 (3.80 Å).
In Remdesivir (D2), two kinds of interactions were predicted between Remdesivir and the MPro(6M03). One is a hydrogen bond, and another is a hydrophobic bond, engaged, respectively, with PHE294 (2.92 Å), ASP293 (3.17 Å), THR111 (2.16 Å), GLN110 (2.94 Å), and ASN131 (3.78 Å), VAL208 (3.78 Å), PRO292 (2.92 Å), VAL 308 (4.52 Å), and PHE305 (5.13 Å), respectively.
Table 3. Protein Ligand interactions of Mpro(PDB ID: 6M03) with top 10 bioactive alkaloid


Drug-likeness properties
Studying drug-like qualities speed up drug discovery and development. One of the main criteria for becoming a drug is Lipinski's rule of five (RO5) [32]. Ten selected alkaloids (L-3, L-7, L-13, L-18, L-25, L-26, L-34, L-39, L-42, and L-49) passed the "RO5" without a single violation (Table 4). The Ghose filter rule, Veber rule, Egan rule, and Muegge rule were also examined for drug-likeness qualities (Table 4). These metrics use their own criteria to select bioactive functions as effective drugs. LogP value: 0.00 to 3.32, molecular weight: 279.3-394.5 g/mol [33]. A ligand should contain ten or fewer rotatable bonds with 100 of the polar surface area, according to the Veber rule [34]. The polar surface area (PSA) and A logP 98 determine a pharmacological molecule's absorption, according to Egan's rule. An effective medicine should pass a pharmacophore point filter to meet the Muegge criterion [35]. The Ghose filter rule, Veber rule, and Egan rule were not violated by our study's alkaloids.
ADME prediction
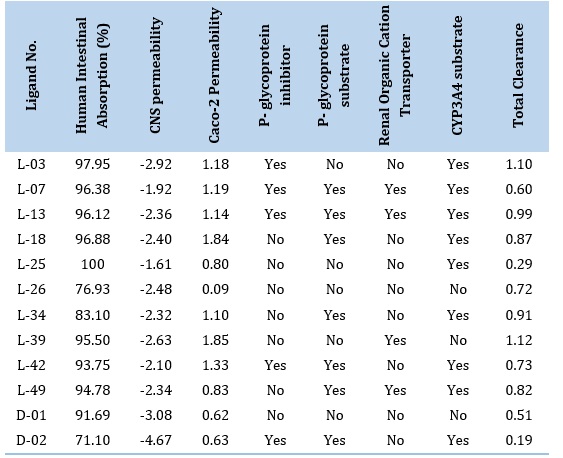
From the ADME study of the top ten docking score compounds (Tables 4 and 5), it is found that hydrogen bond acceptors are 3-7 while donors are 0-2, topological polar surface area (TPSA) ranges from (30.9 to 66.5) Ų. Lipophilicity (XLOGP3) ranges from 0.00 to 3.46. Water solubility (ESOL) ranges from -2.55 to -4.38 and Bioavailability is 0.55. The highest molecular weight (MW) is 365.3, and the lowest is 263.2. The ADME properties of the selected ten compounds are as follows: molecular weight is (365.3, 329.4, 285.3, 394.5, 263.2, and 313.3) g/mol, and the number of hydrogen bond acceptors is 7, 5, 4, 3, and 4. The number of hydrogen bond donors is 0, 1, 2, 0, and 0. TPSA is (66.5, 51.2, 52.9, 51.2, 40.5, and 30.9) for (L-3, L-7, L-13, L-18, L-25, and L-26) respectively. Lipophilicity (XLOGP3) is 3.03, 0.00, 0.00, 3.24, 0.00, and 3.46 for the ligands L-3, L-7, L-13, L-18, L-25, and L-26, respectively. BBB+ signifies the chemical can pass the BBB, and human intestinal absorption (HIA+) is positive, as shown in Table 4. Currently, for COVID-19 the supportive therapy favipiravir and remdesivir have molecular weight 157.1 and 602.6, hydrogen bond acceptors range of 13 and hydrogen bond donor 4. Lipophilicity (XLOGP3) are 0.930 and -0.145. Solubility (ESOL) ranges are -1.65 and -3.473; topological polar surface area (TPSA) ranges are 84.6 and 204. The plasma protein binding is 54-88%.
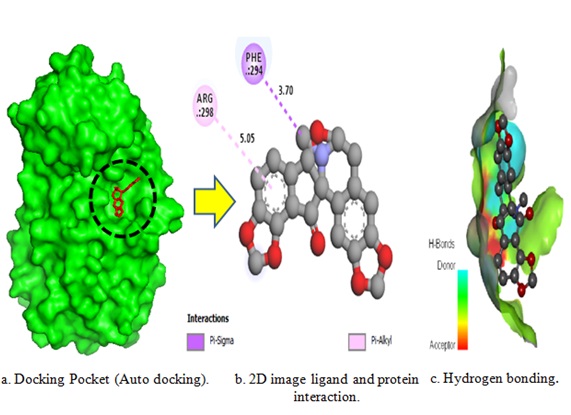


Figure 1. Different docking poses of L-3, L-7 and L-13 sample against MPro (6M03).
Table 4. Pharmacokinetic Parameters of the best 10 alkaloids
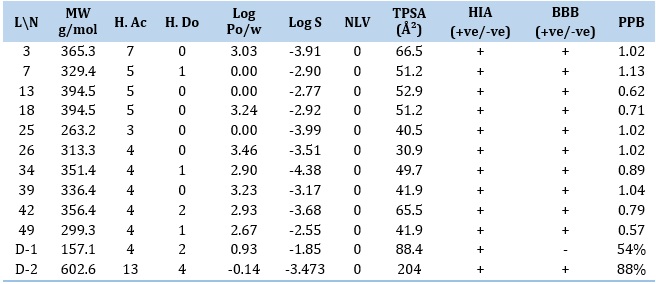
Table 5. Pharmacokinetic parameters of the best docking score compounds
Prediction of toxicological properties of the potential alkaloid
The toxicological features of the chosen alkaloids were also anticipated since toxicity is a key issue when administering any medicine. The toxicological properties of the ten compounds were predicted using the online toolset admetSAR (Table 6). The results represents that all of the alkaloids were likely non-carcinogenic, acute oral toxicity is category III except for 3 and 10, Avian toxicity is negative, and Fish aquatic toxicity is + except for 1. Hepatotoxicity is negative, except for 1 and 5. Biodegradation is negative, and honey bee toxicity is negative except for 2, 6, 8, and 10. The acute oral toxicity category is III, avian toxicity, carcinogenicity, and biodegradation are negatives; and aquatic fish toxicity, and hepatotoxicity are present in favipiravir (D-1) and remdesivir (D-2).
Calculation of quantum properties by DFT
To examine the chemical stability of our target compounds, the electrical characteristics of the top 10 bioactive alkaloid ligands were analyzed using DFT method (Supplementary: ST-4). First of all, HOMO and LUMO energies are evaluated and it has used to derive global reactivity descriptors, such as electrophilicity, chemical potential, electro-negativity, hardness, and softness (S). HOMO and LUMO are important concepts in molecular orbital theory, a framework used in chemistry to understand the electronic structure and reactivity of molecules. These terms stand for the Highest Occupied Molecular Orbital (HOMO) and the Lowest Unoccupied Molecular Orbital (LUMO) [36-38]. Table 7 presents the chemical reactivity of the top 10 bioactive alkaloids, with electron gaps ranging from 0.12 to 3.42 eV and softness and hardness ranging from 0.09 to 1.6 eV and 0.06 to 1.71 eV, respectively. The electro negativity, a measure of an atom's attraction to electrons in a covalent connection, varies from -3.51 to 10.87 eV. The electrophilicity index measures the reduction in total energy caused by the maximum electron flow between donors and acceptors. The electrophilicity varies between 0.13 and 58.54 eV.
Table 6. Steps of the anti-inflammatory activity
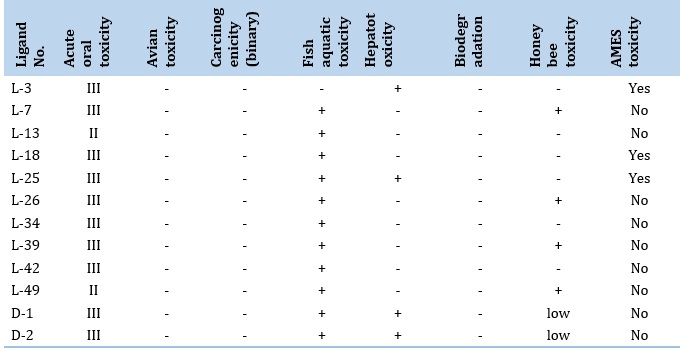
Table 7. Data of quantum properties

Conclusion
The current research shows that alkaloids, which are one of the most common natural substances, show promise as a treatment for COVID-19 because they act on many therapeutic targets at the same time and have strong antiviral effects. As a result of the contents indicated, broad and comprehensive clinical research on these substances seem valuable and important. The new study also explores a larger number of anti-SARS-CoV-2 alkaloids and their molecular mechanisms.The HOMO-LUMO and LUMO-HOMO gaps determine their chemical reactivity as well as their softness and hardness to produce medicines. The pharmacokinetic analysis reveals that they have varied values, yet all medicines can meet the Lipinski's rule of five (RO5). Finally, the ADMET data provides crucial information about a medication and its use in a human cell with relatively minimal toxicity, despite the fact that all pharmaceuticals are non-carcinogenic materials. A molecular docking analysis revealed that all medicines have a high affinity for corona main protease (Mpro). Initially, L-3, L-7, L-13, L-18, and L-25 showed that docking score as binding energy at -8.6, -8.3, -8.2, -8.2, and -8.1 kcal/mol. Compared to the World Health Organization (WHO) guidelines and FDA approved antiviral drug of favipiravir and remdesivir which have binding energies -5.1 and 8.1 kcal/mol of MPro (6M03) and L-3, L-7, L-13, and L-18 have shown binding energies -8.6, -8.3, -8.2, and -8.2 kcal/mol. Thus, these values indicate that the L-3, L-7, L-13, and L-18 candidate molecules will show strong activity against the SARS-CoV-2.
Disclosure Statement
No potential conflict of interest was reported by the authors.
Orcid
Md Abul Hasan Roni : 0000-0003-4912-848
Md. Golam Mortuza : 0000-0002-0019-5704
Rozina : 0009-0001-8973-0119
Ripon Kumar Shaha : 0000-0002-8508-2089
Sajidul Hoque : 0000-0003-3879-5198
Ajoy Kumer : 0000-0001-5136-6166
)